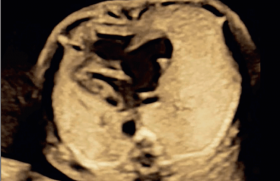
Infertilité
Frontières
Publié le 14 fév 2019Lecture 13 min
Signes d’appel échographiques et ACPA (analyse chromosomique sur puce à ADN)
Valérie MALAN, MD, PhD, service de cytogénétique, Hôpital Necker-Enfants Malades, Université Paris Descartes, Paris, France
*Article écrit durant l'année 2017*
Vers la fin des années 1990, le séquençage du génome humain a permis d’obtenir des fragments d’ADN séquencés qui ont été clonés dans des bactéries ou des levures. Vers la fin des années 1990, Pinkel et al. et Solinas-Toldo et al. ont eu l’idée de déposer sur des lames de verre ces séquences d’ADN dont la position dans le génome était connue(1,2). C’est ainsi que les premières puces à ADN ou « microarray » sont apparues et que la technique de CGH array s’est développée. Actuellement, ce sont des oligonucléotides (ou « sondes ») d'environ 60 bases qui sont fixés sur les lames. Ainsi, en fonction du nombre de sondes choisies, la résolution de la puce peut varier d’environ 1 Mb (mégabase) à 1 kb (kilobase).
Principe de la technique de CGH array
Son principe consiste à cohybrider la même quantité d’ADN provenant d’un patient et d’un témoin, marquées chacune par un fluorochrome différent, sur une lame sur laquelle sont fixées des sondes. Après une étape d’hybridation, les signaux générés par les deux fluorochromes sont numérisés et un rapport de leur intensité respective (reflétant le rapport de la quantité d’ADN du patient par rapport à celle du témoin) est établi au niveau de chaque locus. Une représentation graphique de l’anomalie est obtenue grâce à un logiciel.
Annexe : Principe de la technique de CGH array.
Son principe consiste à cohybrider la même quantité d’ADN provenant d’un patient et d’un témoin, marquée chacune par un fluorochrome différent, sur une lame sur laquelle sont fixées des sondes. Après une étape d’hybridation, les signaux générés par les deux fluorochromes sont numérisés et un rapport de leur intensité respective (reflétant le rapport de la quantité d’ADN du patient par rapport à celle du témoin) est établi au niveau de chaque locus. Une représentation graphique de l’anomalie est obtenue grâce à un logiciel.
Grâce à cette nouvelle technologie, il a donc été possible de détecter des déséquilibres génomiques qui ne sont pas visibles sur le caryotype, c’est-à-dire ayant une taille inférieure à 5-10 Mb (ce qui correspond à la taille d’une bande chromosomique) (figure 1).
Par la suite, d’autres types de puces, dites puces SNP ou « SNP array » (single nucleotide polymorphism), ont été développés. Ce sont des puces qui utilisent des oligonucléotides courts et très spécifiques pouvant distinguer des séquences ne différant que d’un seul nucléotide. En moyenne, un SNP est rencontré tous les 100 à 1 000 nucléotides. Leur nombre est d’environ 5.106 dans le génome humain. Les puces SNP offrent l’avantage de détecter, en plus des déséquilibres génomiques, les situations de disomie uniparentale (deux chromosomes issus d’un même parent). En France, le terme d’analyse chromosomique sur puce à ADN (ACPA) a été choisi par le réseau français « AChro-Puce » pour désigner aussi bien les techniques de CGH (comparative genomic hybridization) array ou puce d'hybridation génomique comparative que les techniques de SNP array (http://www.renapa.univ-montp1.fr/).
Figure 1.
Applications médicales de l’ACPA
Le développement de la technique d’ACPA (ou analyse chromosomique sur puce à ADN) a donc conduit à un changement de paradigme en cytogénétique médicale. Les premières applications de l’ACPA concernaient la cytogénétique des tumeurs solides en raison de la complexité des anomalies détectées sur le caryotype. La pathologie constitutionnelle a été le second champ d’application médicale de cette technique avec l’exploration des patients présentant un déficit intellectuel isolé ou syndromique. Différentes études ont montré que l’ACPA permettait de détecter environ 12 % de déséquilibres génomiques cryptiques (c’est-à-dire non visibles sur le caryotype) chez ces patients(3). C’est pourquoi cette technique est devenue l’examen de première intention pour l’analyse génomique des patients avec déficit intellectuel et/ou malformations congénitales. Cependant, l’impact clinique d’une anomalie identifiée par ACPA peut être parfois difficile à déterminer, constituant un frein majeur à l’application de cette technique en prénatal. Les moyens techniques ont devancé notre compréhension des implications médicales des données générées.
Les CNVs (copy number variants) : classification
Les déséquilibres génomiques mis en évidence par ACPA sont appelés CNVs (copy number variants) et correspondent à des variations quantitatives du génome en comparaison à un génome de référence*.
Il est important de souligner qu’un CNV correspond uniquement à une perte ou un gain de matériel chromosomique sans que l’on puisse préjuger de sa pathogénicité. En effet, un CNV peut être bénin c’est-à-dire sans conséquence phénotypique pour l’individu porteur. Actuellement, on estime qu’environ 16 % du génome correspondraient à des variations non délétères(4). Cependant, il a été montré que certains de ces CNV dits bénins ont un rôle dans l’immunité ou la survenue de pathologie commune comme le lupus systémique(5,6).
À côté de ces CNV bénins, il existe des CNV pathogènes car à l’origine de troubles neuro-développementaux. De nombreux syndromes comme la délétion 17q21.31 (syndrome de Koolen-de Vries) ont pu être ainsi décrits suite à la généralisation de l’ACPA en postnatal(7). Il est à noter que la majorité des CNV pathogènes ne sont pas récurrents et sont dispersés sur l’ensemble des chromosomes. Ainsi, une approche pangénomique et non ciblée est nécessaire pour les détecter.
À côté de ces situations claires, il existe des CNV qui posent des problèmes de conseil génétique. Il s’agit d’une part des CNV de prédisposition à la survenue de pathologies neuro-développementales. C’est par exemple le cas de la délétion 16p11.2 ou de la duplication 1q21.1 qui sont associées à des troubles du spectre autistique(8,9). Des études cas-témoins ont montré que ces CNV constituaient des facteurs de susceptibilité à ces pathologies car ils sont plus fréquemment détectés chez les patients que chez les témoins. Cependant, actuellement, on considère que cette dichotomie entre individus sains et témoins ne reflète pas la réalité, car il existe en fait un spectre phénotypique très large chez les individus porteurs de ce type de CNV. En effet, des troubles cognitifs a minima ont été décelés rétrospectivement chez des parents porteurs alors qu’ils avaient été considérés initialement comme sains(10,11). À ce jour, les facteurs génétiques, épigénétiques et/ou environnementaux qui conduisent à l’expression de la maladie ne sont pas connus. Le conseil génétique est donc très difficile en anténatal car il n’est pas possible de prédire le phénotype d’un fœtus porteur de ce type de CNV.
Par ailleurs, la seconde catégorie de CNV qui soulève des problèmes de conseil génétique correspond aux VUS (variation of uncertain significance, ou variant de signification incertaine). Il s’agit de CNV de caractère « privé » et souvent hérités dont les conséquences cliniques pour le fœtus porteur ne sont pas établies. Cette situation génère une grande anxiété lorsque l’information est transmise aux parents et peut conduire à une demande injustifiée d’interruption médicale de grossesse.
Enfin, de façon exceptionnelle, un CNV prédisposant à la survenue d’une pathologie à révélation tardive peut aussi être mis en évidence(12). L’application de cette nouvelle technologie en prénatal doit ainsi amener à considérer les problèmes éthiques qu’elle soulève.
Du fait de la difficulté d’interprétation de certains CNV (VUS) ou de la possible détection d’un CNV ayant un impact médical sans lien avec l’indication initiale (CNV de découverte fortuite ou « incidental finding »), les recommandations internationales préconisent qu’une consultation pré-test soit proposée à la patiente afin qu’une information précise et claire soit délivrée par un personnel de santé formé (médecin, sage-femme, conseiller en génétique…)(13,14). Le recueil d’un consentement spécifique en vue de la réalisation de cette analyse est indispensable. Enfin, lorsqu’un CNV est notifié sur le compte-rendu, celui-ci doit être expliqué à la patiente lors d’une consultation post-test.
Avantages et limites de l’ACPA en prénatal
Au cours de ces dernières années, la faisabilité et les avantages de l’application de l’ACPA en prénatal ont été largement démontrés(15). Les principaux avantages de cette technique en prénatal sont :
– la possibilité de détecter des CNV pathogènes cryptiques ;
– une détection plus rapide que le caryotype des CNV non cryptiques, car un résultat peut être obtenu en moins d’une semaine du fait que la technique soit effectuée sur des cellules non cultivées (à partir d’un prélèvement de liquide amniotique, de villosités choriales ou de sang fœtal) ;
– la possibilité d’analyser des cellules qui ne sont pas capables de se diviser. Une ACPA peut être réalisée à partir de tissus fœtaux (peau, poumon, thymus..) après une mort fœtale in utero par exemple ;
– une caractérisation fine du déséquilibre génomique identifié (taille du CNV et contenu en gènes). La caractérisation au niveau moléculaire d’un remaniement chromosomique permet d’établir une corrélation phénotype - génotype plus précise et donc de donner un conseil génétique plus adéquat.
Bien que l’ACPA soit une technique très performante, il est important de souligner qu’elle présente des limites. En effet, le principe de la technique étant de comparer la quantité d’ADN d’un patient par rapport à un témoin, les remaniements équilibrés (sans perte ni gain de matériel chromosomique) ne sont pas mis en évidence. Par ailleurs, une analyse cellule par cellule n’est pas possible, contrairement au caryotype ou à la FISH (fluorescence in situ hybridization) et, de ce fait, les faibles mosaïques (< 10-20 %) ne sont pas détectées. Enfin, les triploïdies ne peuvent être décelées que par les puces SNP et non par la CGH array.
Une autre limite à considérer est le fait que l’ACPA donne une information quantitative sans préciser le type de l’anomalie identifiée (dérivé de translocation ou d’insertion par exemple). La mise en évidence d’un CNV devra donc être complétée par une étude FISH. De plus, une étude FISH chez les parents est indispensable pour déterminer le caractère de novo ou hérité de l’anomalie, ce qui aidera, dans certaines situations, à la classification du CNV (pathogène ou non).
Les indications d’ACPA en prénatal
Un diagnostic prénatal chromosomique est proposé lorsqu’il existe une augmentation significative du risque d'anomalie chromosomique déséquilibrée chez le fœtus susceptible d'entraîner des troubles du développement suffisamment graves et/ou létaux pour proposer une interruption médicale de grossesse. Différentes études ont montré que le taux de CNV pathogènes cryptiques détecté par ACPA était d’environ 9 % chez les fœtus polymalformés. En cas de malformation isolée, ce taux est plus faible aux alentours de 6 %(16). Ainsi, la réalisation d’une ACPA pangénomique chez les fœtus présentant une ou des anomalie(s) morphologique(s) est une stratégie diagnostique qui a été adoptée dans de nombreux pays et notamment en France (Recommandations ACLF http://www.eaclf.org/docs/ ACPA/GBP_ACPA-v10.pdf). D’après la métaanalyse de de Wit et al., les malformations cardiaques, du système nerveux central et musculo-squelettiques sont les plus pourvoyeuses d’anomalies chromosomiques pathogènes(16).
L’ACPA chez les fœtus présentant une cardiopathie
Les cardiopathies sont une des malformations congénitales les plus fréquentes à la naissance. Leur prévalence est estimée entre 4 et 8 pour 1 000 naissances vivantes. En anténatal, l’incidence des anomalies chromosomiques chez les fœtus présentant une cardiopathie est élevée, de l’ordre de 25 %(17). Il s’agit essentiellement de cas de trisomie 21, trisomie 18, trisomie 13, monosomie X et microdélétion 22q11.21. Une récente métaanalyse de Jansen et al. incluant 1 131 fœtus présentant une cardiopathie a révélé que l’ACPA permettait de détecter environ 7 % d’anomalies (après exclusion des aneuploïdies et des cas de microdélétion 22q11.21)(18). Ce taux est plus élevé dans les formes syndromiques (9,3 %) que dans les formes isolées (3,4 %). Par ailleurs, une anomalie chromosomique est plus fréquemment retrouvée en cas de communication inter-ventriculaire syndromique et d’anomalies des voies d’éjection ventriculaire gauche. Il est à noter que des CNV pathogènes ont également été identifiés chez des fœtus présentant une transposition des gros vaisseaux ou une hétérotaxie. Actuellement, il est donc recommandé de réaliser une ACPA quel que soit le type de cardiopathie(18-20).
L’ACPA chez les fœtus présentant une anomalie du système nerveux central
Les anomalies de système nerveux central sont souvent sévères et représentent une cause fréquente d’interruption médicale de grossesse(21). Elles concernent 0,14-0,16 % des naissances vivantes et 2-6 % des enfants mort-nés. Le taux de CNV pathogènes chez les fœtus présentant une anomalie du système nerveux central est d’environ 7-11 % selon les études(22,23). Il s’agit d’une des malformations congénitales les plus fréquemment associées à un CNV pathogène. Ces déséquilibres génomiques sont surtout observés chez les fœtus présentant une malformation de Dandy-Walker, une hypoplasie cérébelleuse ou une holoprosencéphalie.
L’ACPA chez les fœtus présentant une hyperclarté nucale
Il a été montré dès le début des années 1990 que l’hyperclarté nucale est associée à un risque d’anomalies chromosomiques comme les trisomies 21, 18 et 13, les triploïdies et les aneuploïdies des gonosomes. Il est à noter que la trisomie 21 représente environ 50 % des anomalies chromosomiques observées en cas d’hyperclarté nucale au premier trimestre(24,25). Plus récemment, plusieurs études ont montré l’apport de l’ACPA dans cette indication(16,23,26). Dans une récente métaanalyse de Grande et al. incluant 1 696 fœtus provenant de 17 études, le taux de CNV pathogène est de 4 % en cas d’hyperclarté nucale isolée et augmente jusqu’à 7 % si un autre signe d’appel échographique est associé(27).
L’ACPA chez les fœtus présentant un retard de croissance intra-utérin
Il est connu que la plupart des anomalies chromosomiques sont associées à un retard de croissance intra-utérin (RCIU). C’est notamment le cas de la trisomie 18, la trisomie 13, la délétion 4p16.3 (syndrome de Wolf-Hirschhorn), la délétion 5p15.2 (syndrome du cri du chat) et la délétion 15q26 (incluant le gène IGF1R)(28). Concernant l’apport de l’ACPA dans cette indication, l’étude de Shaffer et al. a montré qu’un CNV pathogène était retrouvé dans 2,7 % des cas(23). Ce taux augmente à environ 10 % si une malformation congénitale est associée au RCIU. La récente métaanalyse de Borrell et al. révèle des taux similaires avec environ 4 % de CNV pathogènes en cas de RCIU isolé et d’environ 10 % s’il est associé à une malformation congénitale(29). Il est donc tout à fait licite de proposer une ACPA en cas de RCIU inférieur au 3e percentile sans étiologie identifiée, même si aucune autre anomalie morphologique fœtale n’est associée.
Quel type d’ACPA doit-on utiliser en prénatal ?
Le principal frein à l’application de l’ACPA en prénatal est lié à la présence des VUS et des CNV prédisposant à des pathologies dont il est difficile de prédire le phénotype (surtout en l’absence de signes d’appel échographiques, ou si les signes d’appel échographiques sont mineurs). De ce fait, une réflexion s’est imposée sur le type de puce à ADN à utiliser en anténatal : l’objectif était de détecter le maximum de CNV pathogènes tout en limitant la détection des CNV soulevant des problèmes de conseil génétique. Or, il a été montré que le niveau de résolution de la puce utilisée est corrélée à la probabilité de détecter des VUS(30,31). De ce fait, certains centres ont choisi d’adopter une puce différente et/ou avec une autre résolution que celle utilisée en postnatal. En France, une puce pangénomique enrichie sur les régions associées à des syndromes microdélétionnels ou microduplicationnels connus ainsi que sur les gènes impliqués dans des maladies sévères est souvent utilisée(32). Par ailleurs, le seuil de détection d’un CNV a été fixé à 1,5 Mb par la plupart des équipes en France. Dans l’avenir, le développement des bases de données publiques (DECIPHER http://decipher.sanger.ac.uk/ ; Database of Genomic Variants http://projects.tcag.ca/variation/) ou privées permettra de mieux préciser l’impact clinique de certains de ces CNV.
Conclusion
L’avènement des puces à ADN est une avancée considérable dans le domaine de la cytogénétique. En effet, cet outil offre la possibilité de réaliser un examen pangénomique à un très haut niveau de résolution. Cependant, la détection d’un déséquilibre génomique soulève parfois de nombreuses questions quant à ses conséquences cliniques, ce qui constitue une limite majeure à l’application de l’ACPA en situation de diagnostic prénatal. Cependant, l’ACPA est actuellement la technique de choix pour l’analyse du génome fœtal lorsqu’un geste invasif pour étude chromosomique est indiqué.
*Un génome de référence est utilisé pour la CGH array mais pas pour les puces SNP.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :