Gynécologie
Cas cliniques
Publié le 29 oct 2020Lecture 5 min
Imagerie anténatale : quand évoquer le syndrome de Zellweger ? Description de deux cas de diagnostic prénatal
L. Benoit, Y. Athiel, L. Salomon, P. Sonigo, T. Attie-Bitach, Y. Ville, Paris
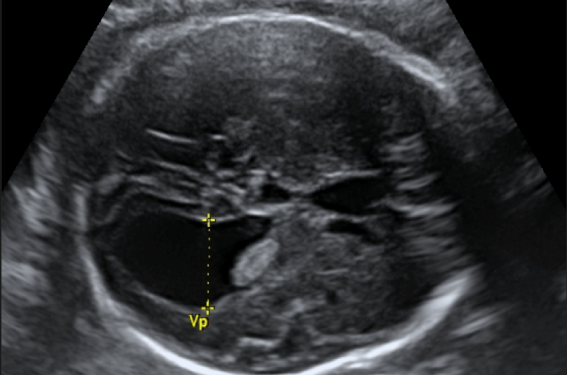
Le syndrome de Zellweger ou syndrome cérébro-hépato-rénal est une maladie métabolique rare, secondaire à une mutation du gène PEX1 nécessaire à la biogenèse des peroxysomes. L’atteinte neurologique est sévère et le pronostic est sombre, avec un décès survenant majoritairement dans la première année de vie. Certains des symptômes sont accessibles à l’imagerie prénatale, mais la rareté du syndrome de Zellweger et l’absence de spécificité des signes échographiques rendent son diagnostic difficile en l’absence de cas index. Il reste cependant possible grâce à la bonne connaissance de l’association des signes échographiques et l’apport de l’IRM. Le syndrome de Zellweger ou syndrome cérébro-hépato-rénal est une maladie métabolique rare, secondaire à une mutation du gène PEX1 nécessaire à la biogenèse des peroxysomes. L’atteinte neurologique est sévère et le pronostic est sombre, avec un décès survenant majoritairement dans la première année de vie. Certains des symptômes sont accessibles à l’imagerie prénatale, mais la rareté du syndrome de Zellweger et l’absence de spécificité des signes échographiques rendent son diagnostic difficile en l’absence de cas index. Il reste cependant possible grâce à la bonne connaissance de l’association des signes échographiques et l’apport de l’IRM.
Le syndrome de Zellweger affecte 1 nouveau-né sur 50 000. Il s’agit d’une maladie à transmission autosomique récessive, dont la cause la plus fréquente est une atteinte du gène de la peroxine PEX1 qui altère la fonction des peroxysomes et prévient la dégradation des acides gras à très longue chaîne (AGLC) qui s’accumulent dans le plasma(1,2).
En cas de grossesse à risque, du fait de l’antécédent d’un cas dans la fratrie, le diagnostic prénatal de syndrome de Zellweger peut être fait après dosage des AGLC, réalisé sur des amniocytes en culture ou des prélèvements de villosités choriales.
Nous décrivons deux cas de syndrome de Zellweger dont le diagnostic avait été évoqué grâce à l’échographie et l’apport de l’IRM prénatale.
Le syndrome de Zellweger est caractérisé par l’association suivante(3,4) :
dysmorphie faciale caractéristique ;
hypotonie néonatale sévère (aréflexie, épilepsie et nystagmus sans aucune acquisition psychomotrice) ;
troubles neurosensoriels (cataracte, choriorétinite, atrophie optique, surdité neurosensorielle et centrale) ;
épilepsie ;
anomalies hépatorénales (kystes hépatiques et rénaux, fibrose, cirrhose) ;
la taille est habituellement normale, mais possibles ponctuations épiphysaires, cryptorchidie chez les garçons ou hypertrophie clitoridienne chez les filles.
Description des cas
Patiente n°1
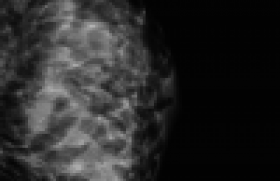
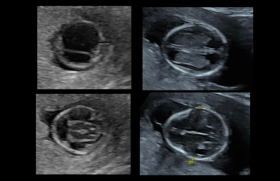
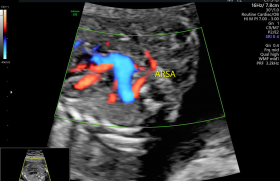
Il s’agissait d’une patiente âgée de 28 ans, 3e geste, primipare sans antécédent. Le couple n’était pas apparenté. Le dépistage de la trisomie 21 n’a pas été effectué et la première échographie réalisée en Turquie n’était pas disponible. L’amniocentèse retrouvait un caryotype normal, une ACPA sans déséquilibre et la recherche de CMV et toxoplasmose était négative. Les signes retrouvés à l’imagerie anténatale sont regroupés dans le tableau 1.
Devant ces anomalies morphologiques, une anomalie du métabolisme peroxysomal type syndrome de Zellweger a été évoquée. Le couple a formulé une demande d’IMG acceptée par le CPDPN et réalisée à 32 SA + 3 j avant les résultats biochimiques pour la recherche de syndrome de Zellweger sur liquide amniotique.
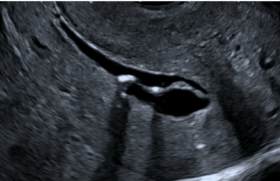
L’examen fœtopathologique a retrouvé une dysmorphie cranio-faciale (front haut, nez large et plat, hypertélorisme, fontanelle antérieure large, yeux légèrement protrus, oreilles basses implantées, basculées en arrière avec hélix repliés, pieds légèrement varus et petits kystes rénaux). L’examen neuropathologique retrouvait foyers de micropolygyrie et d’hétérotopies sous corticales (figure 3), un corps calleux épais et court et des ventricules latéraux larges.
Le diagnostic de syndrome de Zellweger a été confirmé en post-natal par dosage des AGLC sur prélèvement fœtal. Le séquençage du gêne PEX 1 chez les parents a révélé une mutation à l’état hétérozygote. Lors de la grossesse suivante, l’échographie du premier trimestre était sans anomalie et la patiente a bénéficié d’une biopsie de trophoblaste avec recherche spécifique de syndrome de Zellweger qui s’est révélée négative.
Patiente n°2
Cas n° 1
Il s’agissait d’une patiente âgée de 31 ans, primigeste, sans antécédent notable. Les examens d’imagerie sont décrits dans le tableau 2.
Devant ces anomalies morphologiques, une anomalie du métabolisme a été évoquée. Face au pronostic neurologique incertain, le couple a formulé une demande d’IMG acceptée par le CPDPN et réalisée à 30 SA.
L’examen fœtopathologique a retrouvé une dysmorphie cranio-faciale (front haut, ensellure nasale large). L’examen neuropathologique a retrouvé un genou du corps calleux épais, une verticalisation et un élargissement des ventricules latéraux et des hétérotopies au niveau des hémisphères cérébraux et cérébelleux associées à une polymicrogyrie et des anomalies de la substance blanche.
Après avoir été évoqué au regard de l’aspect macroscopique du fœtus, le diagnostic a été posé en post-natal par dosage des AGLC, après relecture des lames histologiques et de l’IRM qui confirmaient a posteriori un aspect compatible avec un syndrome de Zellweger (méthode du « phenotypal reverse »). L’analyse génétique a révélé une mutation du gène PEX 1 à l’état hétérozygote chez les deux parents.
Les grossesses suivantes ont été marquées par deux récidives du syndrome de Zellweger, dont l’une s’est révélée par une hyperclarté nucale à 5,8 mm à l’échographie du premier trimestre et un dépistage de la trisomie 21 retrouvant un risque à 1/12 (ßHCG = 1,26 MoM et PAAP-A = 1,44 MoM) puis une persistance des sacs jugulaires. Le diagnostic a été confirmé après IMG sur le prélèvement anténatal.
Diagnostic anténatal du syndrome de Zellweger
Le diagnostic prénatal du syndrome de Zellweger est possible depuis 1984, depuis les travaux de Moser et coll., qui ont étudié l’accumulation des AGLC dans les peroxysomes sur des amniocytes en culture(5).
Actuellement, la méthode diagnostique utilisée en anténatal reste le dosage des AGLC sur des amniocytes en culture ou des villosités choriales.
Conclusion
Le pronostic néonatal des enfants atteint du syndrome de Zellweger est sombre. Une meilleure connaissance des signes pouvant faire évoquer le syndrome de Zellweger devrait permettre d’améliorer son dépistage en anténatal dans le but de proposer un conseil génétique adapté et, le cas échéant, un diagnostic anténatal pour les grossesses ultérieures.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :